Artykuł opublikowany w czasopiśmie „Chemia w szkole” w 2013 roku. Udostępniono za zgodą redakcji czasopisma.
Karol Dudek, Paweł Bernard „Szkolny eksperyment ilościowy – otrzymywanie gazów” Chemia w szkole 3/2013 s. 5-8
Paca laboratoryjna zajmuje szczególne miejsce w procesie dydaktycznym przedmiotów przyrodniczych. Eksperymenty wykonywane samodzielnie przez ucznia czy prezentowane przez nauczyciela sprawiają, że lekcje są bardziej interesujące [1], a nauczanie i samo uczenie się staje się procesem bardziej efektywnym. Wprowadzona w 2008 roku reforma polskiego systemu oświaty wraz z opracowanymi nowymi podstawami programowymi [2] kładzie duży nacisk na wykorzystanie pracy laboratoryjnej jako jednej z głównych metod nauczania. Zakres treści nauczania stwarza wiele możliwości pracy metodą projektu edukacyjnego (szczególnie o charakterze badawczym), metodą eksperymentu chemicznego lub innymi metodami aktywizującymi, co pozwoli uczniom na pozyskiwanie i przetwarzanie informacji na różne sposoby i z różnych źródeł. Samodzielna obserwacja ucznia jest podstawą do przeżywania, wnioskowania, analizowania i uogólniania zjawisk, stąd bardzo duża rola eksperymentu w realizacji powyższych treści [3].
Nowa podstawa programowa kieruje proces nauczania na stosunkowo nowy tor – samodzielnego dochodzenia ucznia do wiedzy [4], często nazywanego nauczaniem przez odkrywanie lub dociekanie naukowe [5] – IBSE (z j. ang. Inquiry Based Science Education).
Jedna z stosowanych obecnie definicji IBSE brzmi [6]: Dociekanie naukowe to intencjonalny proces polegający na diagnozowaniu problemów, dokonywaniu krytycznej analizy eksperymentów i znajdywaniu alternatywnych rozwiązań, planowaniu badań, sprawdzaniu hipotez, poszukiwaniu informacji, konstruowaniu modeli, dyskusji z kolegami oraz formułowaniu spójnych argumentów.
Alternatywnie można posłużyć się definicją [7]: Uczenie się poprzez odkrywanie: otwarte procesy uczenia się wymagają, by charakteryzować naukę jako aktywne odkrywanie w przeciwieństwie do uczenia się receptywnego. Nie oznacza to, że treść nauczania nie ma być przystępna i zrozumiała. Oznacza to, że sposób nabywania wiedzy lub kompetencji nie może być wyłącznie procesem otrzymywania informacji, lecz zawsze powinien polegać na odkrywaniu.
Zgodnie z założeniami nowej podstawy programowej staje się rzeczą konieczną wzbogacenie lekcji o metody pozwalające uczniowi samodzielnie dochodzić do wiedzy. Poniżej przedstawiono kilka przykładów doświadczeń, które zazwyczaj wykonywane są jedynie podczas omawiania charakterystyki substancji (pierwiastków i substancji gazowych), a tymczasem mogą z powodzeniem zostać wykorzystane jako przedmioty dociekania w zakresie zagadnień dotyczących praw gazowych omawianych w dziale „Obliczenia chemiczne” i do badania kinetyki reakcji chemicznych w dziale „Elementy chemii ogólnej” na IV. poziomie edukacyjnym, poziom rozszerzony.
Doświadczenia dotyczące gazów można wykonać na dwa sposoby, pierwszy ukazujący tylko aspekt jakościowy, drugi scalający elementy ilościowe i jakościowe.
W większości podręczników autorzy proponują klasyczne metody otrzymywania i zbierania substancji gazowych, dostosowane do właściwości otrzymywanej substancji (przykładowo, wodór otrzymuje się w reakcji metalu z kwasem solnym, a powstały gaz zbiera się poprzez wyparcie wody z probówki odwróconej do góry dnem) [8]. Alternatywnie gazy w warunkach szkolnych można otrzymywać i zbierać w układzie zamkniętym, tj. w kolbie stożkowej, na której szyjkę założony został balon [9]. Przed nałożeniem balonu w kolbie umieszczamy odpowiednią ilość roztworu o zadanym stężeniu, natomiast drugi reagent (w fazie stałej) umieszczamy w baloniku. Po nałożeniu balonika przesypujemy jego zawartość do kolby. Podczas reakcji powstaje gaz, który jest zbierany w balonie. Po zakończonej reakcji otrzymany gaz może zostać przepompowany do innych naczyń, w których można przeprowadzić badanie jego właściwości (fot. 1).

Fot. 1. Otrzymywanie gazów w kolbie płaskodennej.
Przykład zastosowania
Otrzymywanie tlenku węgla(IV)
W kolbie stożkowej (nie mniejszej niż 350 ml, możliwie wysokiej) umieścić 60 cm3 12% kwasu chlorowodorowego. W baloniku umieścić ok. 8,9 g stałego węglanu wapnia lub analogiczną ilość rozkruszonych muszelek. Balonik należy nałożyć szczelnie na szyjkę kolby (można dodatkowo zwiększyć szczelność połączenia zabezpieczając gumką recepturką). Wymieszać substraty przesypując zawartość balonika do kolby stożkowej.
Jak wspomniano wcześniej, zebrany gaz można przepompować do innych naczyń w celu przeprowadzenia reakcji charakterystycznych.
Porównanie właściwości wodoru i tlenku węgla(IV)
Doświadczenie przeprowadzić w sposób analogiczny. W baloniku umieścić 2,1 g opiłków magnezu (lub wstążki magnezowej), a w kolbie stożkowej umieścić 60 cm3 12% kwasu chlorowodorowego. Nałożyć balonik i połączyć substraty. Tym razem balonik zawiązany nitką wzlatuje do góry.
UWAGI:
- Ze względu na żrące właściwości kwasu niezbędna jest praca w okularach i rękawicach ochronnych!
- Przeprowadzić reakcję pod dygestorium.
- Na czas doświadczenia wyłączyć wszystkie źródła otwartego ognia.
- Nie podpalać balonika! Po zakończonym doświadczeniu wypuścić otrzymany gaz do atmosfery pod sprawnie działającym wyciągiem.
- Zastosowanie balonu z grubej gumy może utrudnić jego na pompowanie i uniemożliwić przeprowadzenie właściwych obserwacji. Pozostałą ilość gumy po związaniu balonika należy odciąć. Alternatywnie można zastosować prezerwatywy zamiast zwykłych balonów – prowadzi to do otrzymania powtarzalnych, poprawnych wyników.
Opisane doświadczenia mają charakter jakościowy. W podejściu ilościowym gaz musi być zbierany w naczyniu z podziałką miarową. Taką funkcję może również pełnić np. strzykawka połączona z kolbą stożkową, w której prowadzona jest reakcja [10] (rys. 1).
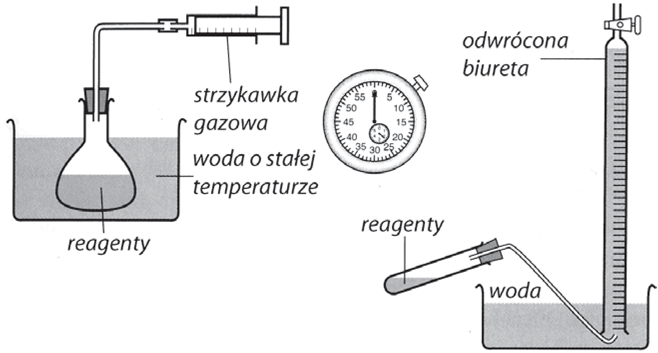
Rys. 1. Schematy układów do otrzymywania gazów
Układ ten można uprościć, przeprowadzając reakcję bezpośrednio w strzykawce lub w próbniku tłokowym z zaworem kapilarnym (fot. 2). Próbniki tłokowe najlepiej sprawdzają się do prowadzenia takich reakcji, niestety szkło tego typu jest dość drogie. Tanią alternatywę stanowi zwykła strzykawka zatkana z jednej strony korkiem.

Fot. 2. Otrzymywanie gazów w próbniku.
Trudność może sprawiać umieszczenie substratów w strzykawce, z tego powodu zamieszczamy szczegółową instrukcję postępowania:
- wyciągamy całkowicie tłok strzykawki (rozłączamy tłok od korpusu),
- zatykamy szczelnie wylot strzykawki, wykorzystać można korek gumowy z naciętym otworem,
- strzykawkę ustawiamy w pozycji pionowej i wypełniamy ją wodą,
- w nakrętce lub innym możliwie płaskim pojemniku (o średnicy umożliwiającej swobodne prze- mieszczanie się po wnętrzu strzykawki) umieszczamy reagent występujący w stałym stanie skupienia,
- nakrętkę umieszczamy na szczycie strzykawki tak, aby unosiła się na tafli wody,
- odtykamy strzykawkę w ten sposób, aby spadek poziomu wody powodował powolne opadanie nakrętki wraz z reagentem na dno strzykawki,
- gdy zakrętka opadnie na dół strzykawki, wprowadzamy tłok i również przesuwamy go w dół, następnie końcówkę strzykawki zanurzamy w reagencie występującym w ciekłym stanie skupienia i pobieramy jego odpowiednią objętość, którą można odmierzyć wykorzystując po- działkę na strzykawce,
- zatykamy ponownie wylot strzykawki,
- obracamy strzykawkę w taki sposób, aby zmieszać substraty reakcji,
- powstający gaz wypycha tłok strzykawki,
- z podziałki odczytujemy objętość wydzielonego gazu.
Eksperyment można przeprowadzić na dwa sposoby:
-
- Uczeń może postępować według instrukcji przegotowanej przez nauczyciela. Instrukcja taka może zawierać dokładny opis czynności laboratoryjnych wraz z ilością wykorzystywanych substratów, stężeniami itd. Eksperyment w takiej formie może być wykonany przed omówieniem prawa Avogadra, a sformułowanie tego prawa na- stępuje na podstawie otrzymanych przez uczniów wyników.
- Uczeń na podstawie równania reakcji, znajomości prawa Avogadra oraz opisanej techniki otrzymywania gazów sam proponuje, jakie parametry będą badane. Przykładowo analizie może zostać poddana wydajność zachodzącej reakcji, kinetyka procesu, stosowalność prawa Avogadra.
Poniżej przedstawiono przepisy pozwalające na otrzymanie kilku podstawowych gazów z wykorzystaniem opisanej techniki. Wartości podane przy każdym doświadczeniu są dostosowane do strzykawki lub próbnika tłokowego o pojemności 100 cm3. Przedstawione proporcje pozwalają otrzymać 80 cm3 gazu zakładając 100% wydajność w warunkach normalnych. Wartości mają charakter orientacyjny i pozwalają na oszacowanie, czy ilości wyliczane przez uczniów są poprawne i realne do zastosowania.
| Doświadczenie 1. Otrzymywanie tlenu
Równanie zachodzącej reakcji: 2H2O2 Masa MnO2: 0,75 g Objętość wody utlenionej (3%): 9 cm3 Komentarz: Uczeń może wykonać to doświadczenie do badania np.: zależność szybkości reakcji od stopnia rozdrobnienia i ilości MnO2 lub stężenia roztworu nadtlenku wodoru. |
 2H2O + O2
2H2O + O2|
Doświadczenie 2. Otrzymywanie chloru 2KMnO4 + 16 HCl → 2 KCl + 5Cl2 + 8H2O + 2MnCl2 Masa KMnO4: 0,23 g Objętość HCl o stężeniu 2 mol/dm3: 15cm3 Komentarz: Doświadczenie można wykonywać nie tylko na lekcjach związanych z obliczeniami, ale także przy omawianiu właściwości chloru, właściwości związków manganu, czy procesów utleniania i redukcji. Uczniowie przed wykonaniem doświadczenia mają za zadanie dobranie odpowiednich współczynników stechiometrycznych, mogą to uczynić za pomocą bilansu elektronowego lub metodą jonowo-elektornową. |
| Doświadczenie 3. Otrzymywanie etynu
CaC2 + 2HCl → CaCl2 + C2H2 Masa CaC2: 0,23 g Objętość HCl o stężeniu 1 mol/dm3: 10 cm3. Efektywność reakcji może być porównana z reakcją zachodzącą pod wpływem wody. CaC2 + H2O → Ca(OH)2 +C2H2 Masa CaC2: 0,23 g Objętość H2O: 10 cm3 Komentarz: Doświadczenie może być nie tylko obrazem praw gazowych, może również stanowić wstęp do lekcji o węglowodorach nienasyconych, badania ich właściwości fizycznych i chemicznych. |
|
Doświadczenie 4. Otrzymywania siarkowodoru [11] ZnS + 2HCl → H2S + ZnCl2 Masa ZnS: 0,33 g Objętość HCl o stężeniu 2 mol/dm3: 10 cm3. Komentarz: Siarkowodór posiada bardzo nieprzyjemny zapach i jest silnie toksyczny. Zaletą przedstawionej techniki jest wykorzystanie układu zamkniętego, zmniejsza to ryzyko uwolnienia się gazu. Fakt ten ma również duże znaczenie w przypadku reakcji otrzymywania chloru. UWAGA. Siarkowodór jest silnie toksyczny. Doświadczenie należy wykonywać pod sprawnie działającym wyciągiem. Otrzymany gaz uwolnić pod wyciągiem do atmosfery. Unikać wdychania gazu. |
Opisane doświadczenia mogą zastosowanie podczas realizacji zagadnień zawartych w nowej podstawie programowej przedmiotu chemia na IV. etapie edukacyjnym – zakres rozszerzony. Podane wymagania szczegółowe wskazują:
Uczeń:
- 1.5 dokonuje interpretacji jakościowej i ilościowej równania reakcji w ujęciu molowym, masowym i objętościowym (dla gazów);
- 1.6 wykonuje obliczenia z uwzględnieniem wydajności reakcji i mola dotyczące: mas substratów i produktów (stechiometria wzorów i równań chemicznych), objętości gazów w warunkach normalnych.
- 4.5 przewiduje wpływ: stężenia substratów, obecności katalizatora, stopnia rozdrobnienia substratów i temperatury na szybkość reakcji; planuje i przeprowadza odpowiednie doświadczenia;
- 8.7 projektuje i przeprowadza doświadczenia pozwalające otrzymać tlen w laboratorium (np. reakcja rozkładu H2O2 lub KMnO4).
Stosowanie nauczania przez do- ciekanie naukowe znajduje coraz szersze zainteresowanie wśród polskich nauczycieli. Zainteresowanie owe jest tym bardziej uzasadnione, że polski system edukacji, wzorując się na zaleceniach Komisji Europejskiej i raportu Rocarda [12], wdraża elementy nauczania przez odkrywanie do polskiej szkoły. W założeniu zmiany te powinny wpłynąć na wzrost zainteresowania uczniów przedmiotami przyrodniczymi oraz na liczbę studentów na kierunkach przyrodniczych, matematycznych i inżynieryjnych. Kwestia słuszności i efektywności podjętych reform nie jest przedmiotem powyższych rozważań, w pracy dydaktycznej jednak nigdy nie należy zapomnieć, że „lepsze jest doświadczenie bez teorii niż teoria bez doświadczenia”.
Literatura:
[1] Burewicz A., Gulińska H., Dydaktyka Chemii, Wydawnictwo UAM, Poznań 2002
[2] Podstawa programowa z komentarzami, tom 5. Edukacja przyrodnicza w szkole podstawowej, gimnazjum i liceum. Załącznik do rozporządzenia Ministra Edukacji Narodowej z 23 grudnia 2008 roku w sprawie pod- stawy programowej wychowania przedszkolnego oraz kształcenia ogólnego w poszczególnych typach szkół z komentarzem
[3] Bernard P., Maciejowska I., Odrowąż E., Dudek K., Geoghegan R., Introduction of inquiry based science education into Polish science curriculum – general findings of teachers’ attitude, Chemistry-Didactics-Ecology-Metrology 2013, Vol. 17 (1–2), s. 49–59
[4] Okoń W., Wprowadzenie do dydaktyki ogólnej, PWN, 1987
[5] Bernard P., Białas A., Broś P., Ellermeijer T., Kędzierska E., Krzeczkowska M., Maciejowska I., Odrowąż E., Szo- stak E., Podstawy metodologii IBSE, [w:] Nauczanie przedmiotów przyrodniczych kształtujące postawy i umiejętności badawcze uczniów, red. Odrowąż E., Maciejowska I., Kraków 2012
[6] Linn M.C., Davis E.A., Bell P., Internet Environments for Science Education, Lawrence Erlbaum Associates, Inc., Mahwah, NJ, 2004
[7] Biuletyn projektu AQUEDUCT, „Nabywanie kluczowych kompetencji poprzez edukację na rzecz dziedzictwa kulturowego,” http://the-aqueduct.eu/download/Aqueduct-Manual_PO.pdf (Ostatni dostęp: 22.05.2012)
[8] Poźniczek M.M., Kluz Z., Wybieram Chemię, t.1, Kraków 2007
[9] Gulińska H., Kuśmierczyk K., Po prostu chemia, Warszawa 2012, s. 10
[10] Brown C., Ford M., Standard Level Chemistry for the IB Diploma, Pearson Baccalaureate 2008
[11] Ibanez G. J., Esparza-Hernandez M., Serrano-Doria C., Infante-Fregoso A., Singh M.M., Environmental Che- mistry. Microscale Laboratory Experiments., Springer 2008
[12] Rocard M., Csermely P., Jorde D., Lenzen D., Walberg-Henriksson H., Hemmo V., Science Education Now: A Rene- wed Pedagogy for the Future of Europe. Brussels, European Communities 2007